Discussion
1. Comaprison results with current tools
2. Parameters optimization and benchmark
1.Comparison results with current tools
Currently, there are no similar tools available specifically for spliced alignment. Although tophat and splicemap could be used for junction site detection, it is hard to know the format intermediate files. However,we have got the 'good' file format from SpliceMap sourcecode and use it to compare with ABMapper.
SpliceMap version:3.11, use default parameter with eland aligner; Tophat version:1.0.13 ABMapper version:2.0, use overlap 16 with all hits output:-multi=0
Hardware:Linux machine is Intel Core2 Duo 2.93GHz, 8GB RAM, Ubuntu 9.10 (OS)
Benchmark criteria:
Accuracy = PPV(%read) =perfect hit / total hit read
PPV(%record) = perfect hit / total hit records
Recall = Sensitivity = perfect hit /total read
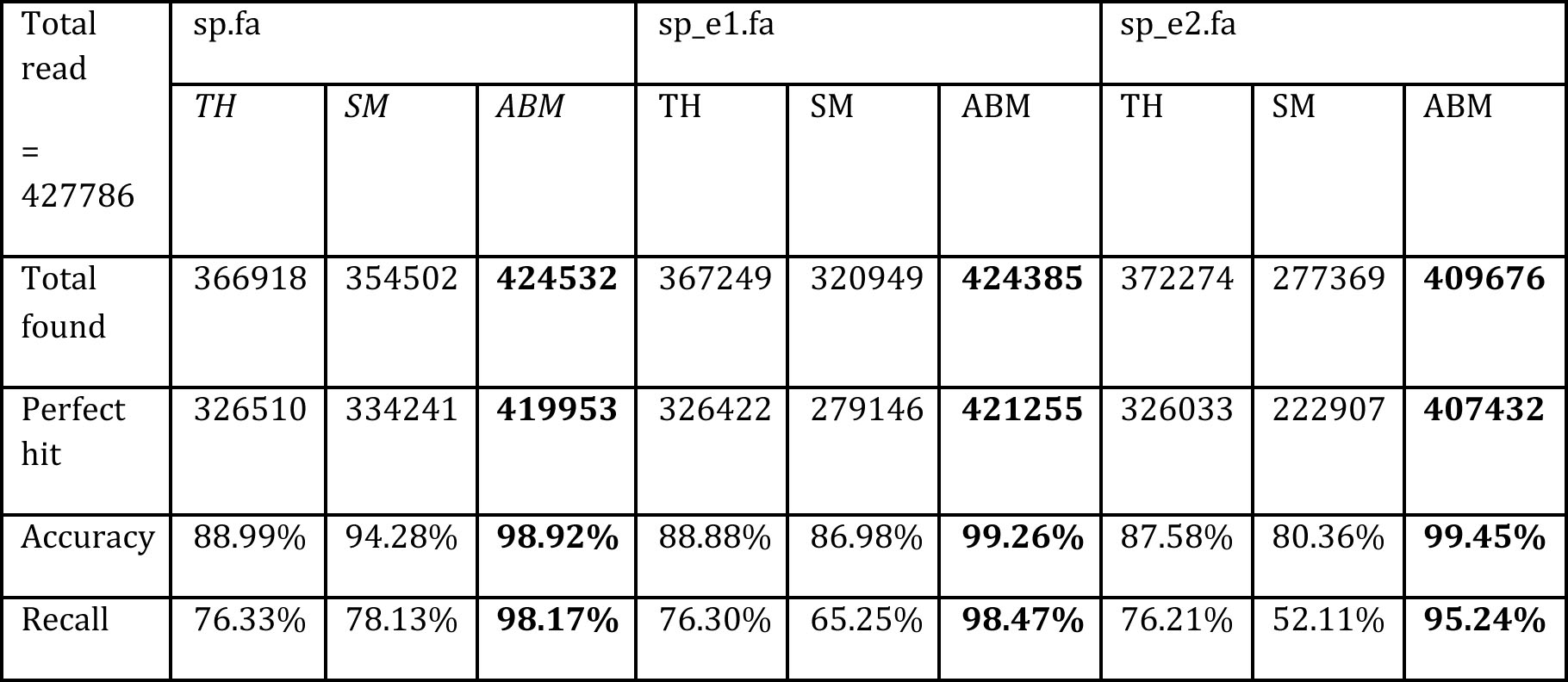
Table 1. Comparison results between TopHat (TH), SpliceMap(SM), and ABMapper (ABM). sp.fa is the benchmark dataset, from which we generate sp_e1.fa and sp_e2.fa placing 1 and 2 random errors respectively. Parameter settings: seed length '-seed'= 11, maximum output '-multi' = 500, and output format '-type' = 1.
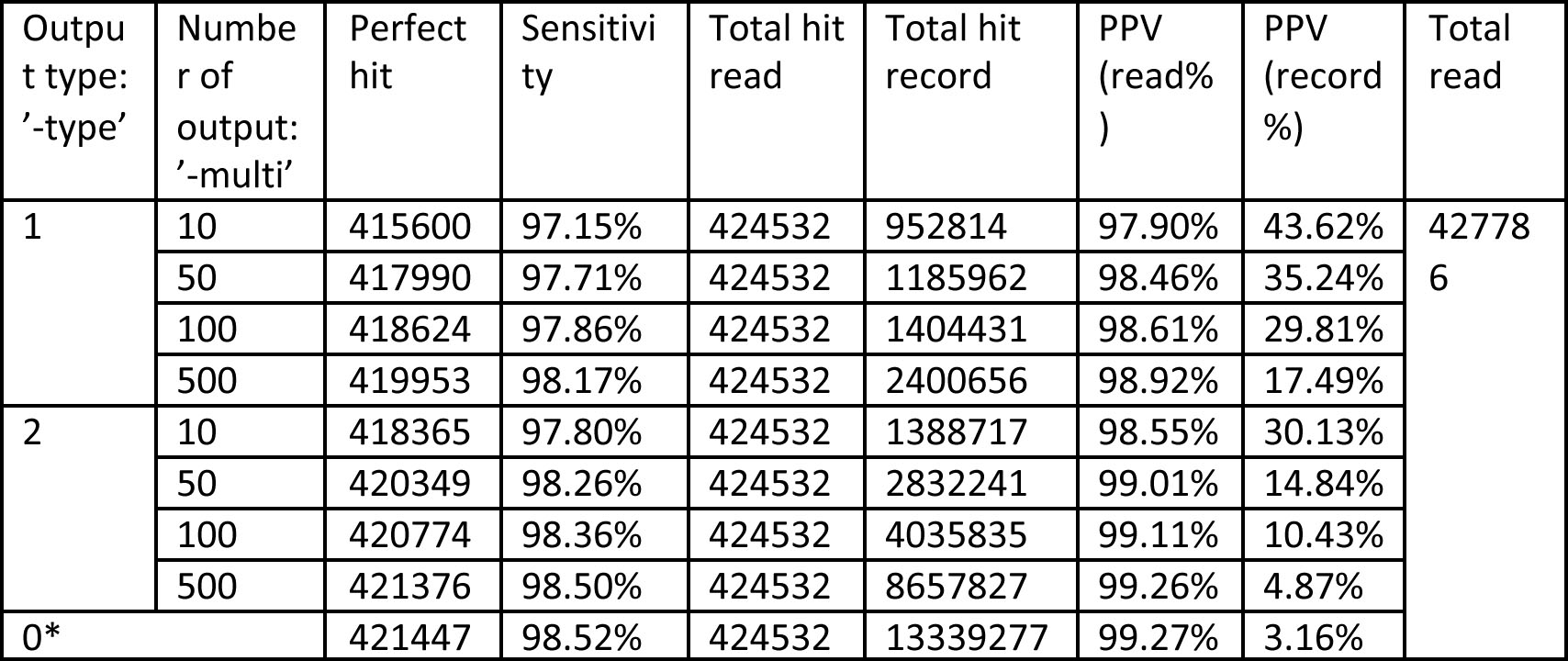
Table 2. Benchmark data (sp.fa) results for different numbers of occurrences output, where seed length ‘-seed’= 11 (*When output is specified as ‘-multi’= 0, all the occurrences are being output.)
An important parameter in ABMapper is 'overlap'. We have also done another analysis to test the 'overlap' parameter(other parameters: -multi 100, -input sp.fq, genome is hg18,total 427786 reads)
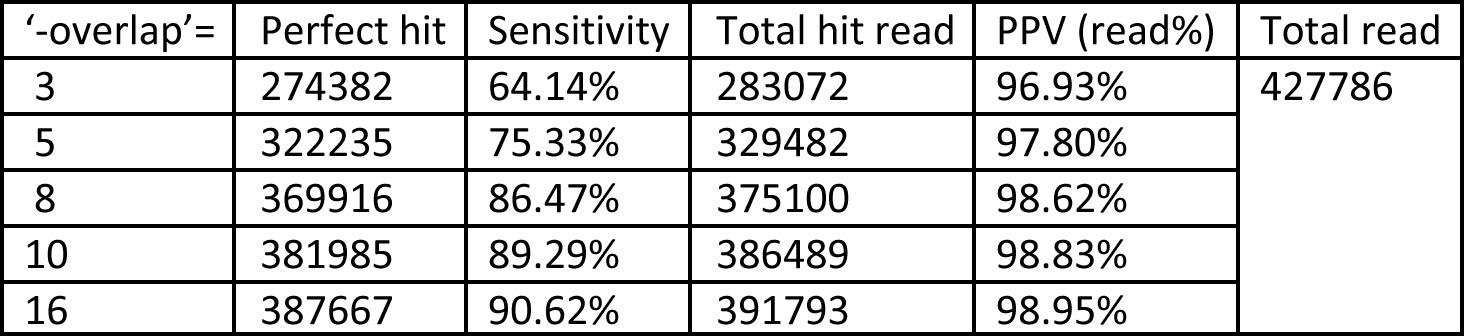
Table 3.Benchmark dataset (sp.fa) with different overlaps, where O = 3, 5, 8, 10, and 16 respectively; seed length ‘-seed’= 11, maximum output ‘-multi’ = 500, and output format ‘-type’ = 1.
2.Parameters optimization and benchmark
1. '-overlap' value is important. most of reads could be over-extended, if there are no overlapping tolerance in spliced alignment It will introduce more False Negative, however, if you set this value high, it would introduce False Positive.Generally, 8 is OK.
2. distance parameters: -min_dist and -max_dist. These two determine the searching range of two seeds hit. If you set it too high, more results and high False Positive error would be introduced. Default is 10-400k
3. '-multi': maximum hits include exonic alignment and 'good' spliced alignment. If you set it too high, huge disk space was required. Default is 500. If you set is 0, it would search all putative alignments.
Fig1. Benchmark by sp.fq(converted from hg18ens.sp.fa,id file for sq.fq which record each read location by order).Hardware configuration is same as above.
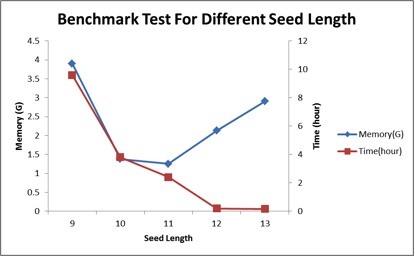
Fig 1. Benchmark (sp.fa) results for different seed length, where maximum output ‘-multi’ = 500, and output type ‘-type’ = 1.
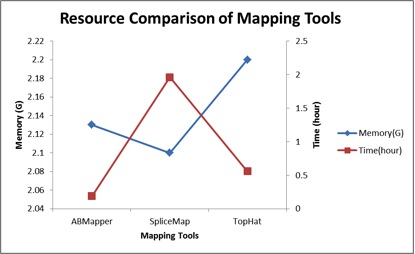
Fig 2. Resource Comparison of Mapping Tools. SpliceMap, TopHat and ABMapper were used to map sp.fa to reference genome (hg18). Eland and Bowtie were used as aligner in SpliceMap and TopHat repectively, ‘-r’ in TopHat was set to 250, other parameters were set as default; Seed length used in ABMapper was 12, maximum output ‘-multi’=500, output type ‘-type’=1;
3.Q & A
Q:1) What about the memory usage on different seeds?
A: if the seed length is 9, the peak memory is about 1.9G; seed length is 10, it about 3G; and if the seed length is 11, the memory need about >10G. So we set 10 as default seed length
Q:2) Does ABMapper support multi-core or pthread?
A: No, We will add this isn the future.
Q:3) Have you compared ABMapper with BWA and Bowtie?
A: No, original idea of ABMapper is different with BWA etc, but user could make use of the advantages of them and ABMapper could also take BWA's SAM file as input.
Q:4) Is ABMapper fast enough?
A: We could get more information at the expense of speed. Details could be estimated by benchmark testing.